
ANZUPGO 20 mg-g, crème, boîte de 1 tube de 60 g
Dernière révision : 19/09/2024
Taux de TVA : 2.1%
Laboratoire exploitant : LEO PHARMA
Source :

Anzupgo est indiqué dans le traitement de l'eczéma chronique des mains (ECM) modéré à sévère chez les adultes pour lesquels les dermocorticoïdes sont inadéquats ou inappropriés (voir rubrique Propriétés pharmacodynamiques).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Cancer de la peau non-mélanome
Des cas de cancer cutané non-mélanome (CCNM), essentiellement des carcinomes basocellulaires, ont été rapportés chez des patients traités par des inhibiteurs de JAK en application topique. Un examen régulier de la peau au site d'application est recommandé chez tous les patients, particulièrement chez ceux présentant des facteurs de risque de cancer cutané.
Excipients à effet notoire :
Alcool benzylique
Ce médicament contient 10 mg d'alcool benzylique (E 1519) par gramme de crème.
L'alcool benzylique peut provoquer des réactions allergiques ou une légère irritation locale.
Butylhydroxyanisole
Le butylhydroxyanisole (E 320) peut provoquer des réactions cutanées locales (par exemple dermatite de contact) ou une irritation des yeux et des muqueuses.
Alcool cétostéarylique
L'alcool cétostéarylique peut provoquer des réactions cutanées locales (par exemple dermatite de contact).
Résumé du profil de sécurité
Les effets indésirables les plus fréquents étaient des réactions au site d'application (1,0 %).
Tableau des effets indésirables
Le Tableau 1 énumère les effets indésirables observés dans les études cliniques. Ces effets indésirables sont présentés par classe de systèmes d'organes MedDRA et par ordre de fréquence selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000).
Tableau 1 Effets indésirables
|
Classe de systèmes d'organes |
Fréquence |
Effet indésirable |
|
Troubles généraux et anomalies au site d'administration |
Fréquent |
Réactions au site d'application* |
* Voir Description de certains effets indésirables
Description de certains effets indésirables
Réactions au site d'application
Dans l'ensemble des trois études cliniques ayant comparé delgocitinib au véhicule de la crème durant 16 semaines, 9 réactions au site d'application (y compris douleur au site d'application, paresthésie au site d'application, prurit au site d'application et érythème au site d'application) ont été rapportées chez 1,0 % des patients traités par le delgocitinib crème. 8 réactions au site d'application étaient d'intensité légère et 1 d'intensité modérée. 7 des 9 réactions sont survenues au cours de la première semaine de traitement. Aucune réaction au site d'application n'a entraîné d'interruption du traitement et le délai médian de résolution a été de 3 jours.
Le taux de réactions au site d'application dans l'étude d'extension à long terme (0,56 événement pour 100 patients-année d'observation) a été inférieur à celui observé au cours des études cliniques de 16 semaines ayant comparé delgocitinib au véhicule de la crème (4,11 événements pour 100 patientsannée d'observation).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE régulier de la peau au site d'application chez tous les patients.
- En cas de réapparition des signes et symptômes de l'ECM (poussées), le
traitement deux fois par jour des zones affectées doit être repris
selon les besoins.
- Le traitement doit être interrompu si aucune amélioration n'est constatée après 12 semaines de traitement continu.
- Si une autre personne applique la crème au patient, elle doit se laver les mains après l'application.
- Le contact avec les yeux, la bouche ou d'autres muqueuses doit être
évité. En cas de contact avec les muqueuses, rincer abondamment à l'eau.
Grossesse
Il n'existe pas de données ou il existe des données limitées (moins de 300 grossesses) sur l'utilisation du delgocitinib chez la femme enceinte.
Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la reproduction (voir rubrique Données de sécurité préclinique).
Par mesure de précaution, il est préférable d'éviter l'utilisation d'Anzupgo pendant la grossesse.
Allaitement
Aucun effet sur le nouveau-né/nourrisson allaité n'est attendu dans la mesure où l'exposition systémique de la femme qui allaite au delgocitinib est négligeable (voir rubrique Données de sécurité préclinique).
Anzupgo peut être utilisé pendant l'allaitement.
Lorsque Anzupgo est utilisé pendant l'allaitement, il convient d'éviter tout contact direct avec le mamelon ou la zone environnante après l'application de la crème sur les mains et/ou les poignets.
Par mesure de précaution, il convient d'éviter tout contact cutané direct lorsqu'on s'occupe d'un nourrisson immédiatement après l'application d'Anzupgo sur les mains et/ou les poignets.
Fertilité
Il n'y a pas de données portant sur l'effet du delgocitinib sur la fertilité humaine.
Sur la base des résultats obtenus chez les rats femelles, l'administration orale du delgocitinib a entraîné une réduction de la fertilité à des expositions considérées comme suffisamment supérieures à l'exposition humaine (voir rubrique Données de sécurité préclinique).
Les études effectuées chez l'animal n'ont pas mis en évidence d'effets sur la fertilité des mâles.
Aucune étude d'interaction n'a été réalisée avec le delgocitinib administré par voie topique ou systémique (voir rubrique Propriétés pharmacocinétiques pour les études d'interaction in vitro). Compte tenu du métabolisme limité du delgocitinib, de l'application sur une surface corporelle limitée (mains et poignets) et de l'exposition systémique minimale du delgocitinib appliqué par voie topique, le risque d'interaction avec les traitements systémiques est faible.
L'utilisation du delgocitinib en association avec d'autres médicaments topiques n'a pas été évaluée et une application concomitante sur les mêmes zones cutanées n'est pas recommandée.
Le traitement par Anzupgo doit être instauré et supervisé par des médecins expérimentés dans le diagnostic et le traitement de l'eczéma chronique des mains.
Posologie
Une fine couche d'Anzupgo doit être appliquée deux fois par jour sur la peau affectée des mains et des poignets jusqu'à ce que la peau ne présente aucune lésion ou presque aucune lésion (voir rubrique Propriétés pharmacodynamiques). Il est recommandé d'appliquer la crème à intervalles réguliers, à environ 12 heures d'intervalle.
En cas de réapparition des signes et symptômes de l'ECM (poussées), le traitement deux fois par jour des zones affectées doit être repris selon les besoins.
Le traitement doit être interrompu si aucune amélioration n'est constatée après 12 semaines de traitement continu.
Oubli de dose
Si l'application d'une dose a été oubliée, la crème doit être appliquée dès que possible. Les applications doivent ensuite reprendre selon le schéma habituel prévu.
Populations particulières
Patients âgés
Aucune adaptation de dose n'est recommandée chez les patients âgés.
Insuffisance rénale et hépatique
Aucune étude avec Anzupgo n'a été menée auprès de patients atteints d'insuffisance hépatique ou rénale sévère. Néanmoins, compte tenu de l'exposition systémique minimale du delgocitinib appliqué par voie topique, aucune adaptation de dose n'est recommandée (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité d'Anzupgo chez les enfants et adolescents de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Anzupgo est réservé à une administration par voie cutanée. Une fine couche d'Anzupgo doit être appliquée deux fois par jour sur la peau propre et sèche des lésions des mains et des poignets. Les patients doivent éviter d'appliquer d'autres produits topiques immédiatement avant et après l'application d'Anzupgo (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). L'application concomitante d'émollients dans les 2 heures précédant et suivant l'application du delgocitinib n'a pas été étudiée.
Si une autre personne applique la crème au patient, elle doit se laver les mains après l'application.
Le contact avec les yeux, la bouche ou d'autres muqueuses doit être évité. En cas de contact avec les muqueuses, rincer abondamment à l'eau.
Durée de conservation :
3 ans
Après la première ouverture : 1 an
Précautions particulières de conservation :
Ne pas congeler.
Sans objet.
Compte tenu de l'absorption systémique minimale du delgocitinib, aucun signe systémique de surdosage n'est attendu après l'application topique d'Anzupgo. Si une quantité excessive de crème a été appliquée, l'excédent peut être essuyé.
Classe pharmacothérapeutique : Autres préparations dermatologiques, agents de la dermatite sauf corticoïdes, Code ATC : D11AH11
Mécanisme d'action
Le delgocitinib est un inhibiteur pan-Janus kinase (JAK) qui cible l'activité des quatre membres de la famille des enzymes JAK, à savoir JAK1, JAK2, JAK3, ainsi que la tyrosine kinase 2 (TYK2), en fonction de la concentration.
Dans les cellules humaines, l'inhibition de la voie JAK-STAT par le delgocitinib réduit la signalisation de plusieurs cytokines pro-inflammatoires (y compris l'interleukine (IL)-2, IL-4, IL-6, IL-13, IL-21, IL-23, le facteur stimulant les colonies de granulocytes et de macrophages (GM-CSF) et l'interféron (IFN)-α), ce qui régule négativement les réponses immunitaires et inflammatoires dans les cellules impliquées dans la pathologie de l'ECM.
Effets pharmacodynamiques
Une étude approfondie de l'intervalle QT réalisée chez des sujets sains n'a pas mis en évidence d'effet d'allongement de l'intervalle QTc du delgocitinib administré par voie orale à des doses uniques allant jusqu'à 12 mg (environ 200 fois l'exposition humaine après application topique, sur la base de la Cmax). Par conséquent, on ne s'attend pas à ce qu'Anzupgo affecte la repolarisation cardiaque dans les conditions d'utilisation clinique.
Études de sécurité cutanée
Les études cliniques réalisées chez des sujets sains ont démontré que le delgocitinib crème n'entraîne pas de réaction cutanée phototoxique ni de réaction cutanée photoallergique.
Efficacité et sécurité cliniques
La sécurité et l'efficacité du delgocitinib crème ont été évaluées dans le cadre de deux études pivots randomisées, en double aveugle, ayant comparé delgocitinib au véhicule de la crème et de méthodologie similaire (DELTA 1 et DELTA 2). L'ECM était défini comme un eczéma des mains persistant pendant plus de 3 mois ou réapparaissant deux fois ou plus au cours des 12 derniers mois. Les études ont inclus 960 patients âgés de 18 ans et plus, atteints d'un ECM modéré à sévère, défini par un score de 3 ou 4 (modéré ou sévère) selon l'évaluation globale par l'investigateur de l'eczéma chronique des mains (Investigator's Global Assessment for Chronic Hand Eczema, IGA-CHE) (voir Tableau 2), et qui devaient présenter un score ≥ 4 points à l'inclusion sur l'évaluation du prurit suivant le Journal des symptômes d'eczéma des mains (Hand Eczema Symptom Diary, HESD) à l'inclusion. Les patients éligibles étaient ceux ayant déjà présenté une réponse inadéquate aux dermocorticoïdes ou pour lesquels les dermocorticoïdes n'étaient pas recommandés (par exemple, en raison d'effets secondaires importants ou de risques pour la sécurité).
Tableau 2 Investigator's Global Assessment for chronic hand eczema (IGA-CHE)
|
Sévérité sur l'IGA- CHE |
Score de l'IGA- CHE |
Signe et intensité |
|
Aucune lésion |
0 |
Absence d'érythème, de desquamation, d'hyperkératose/de lichénification, de vésicules, d'œdème ou de fissures |
|
Presque aucune lésion |
1 |
Érythème à peine perceptible Absence de desquamation, d'hyperkératose/de lichénification, de vésicules, d'œdème ou de fissures |
|
Léger |
2 |
Au moins un des signes suivants : • Érythème léger mais net (rose) • Desquamation légère mais nette (principalement squames fines) • Hyperkératose/lichénification légère mais nette et au moins un des signes suivants : • Vésicules éparses, sans érosion • Œdème à peine palpable • Fissures superficielles |
|
Modéré |
3 |
Au moins un des signes suivants : • Érythème clairement perceptible (rougeâtre) • Desquamation clairement perceptible (squames épaisses) • Hyperkératose/lichénification clairement perceptible et au moins un des signes suivants : • Vésicules groupées, sans érosion visible • Œdème net • Fissures nettes |
|
Sévère |
4 |
Au moins un des signes suivants : • Érythème prononcé (rouge foncé ou vif) • Desquamation prononcée et épaisse • Hyperkératose/lichénification prononcée et au moins un des signes suivants : • Densité élevée de vésicules, avec érosions • Œdème prononcé • Une ou plusieurs fissures profondes |
Dans les études DELTA 1 et DELTA 2, les patients ont appliqué soit le delgocitinib 20 mg/g en crème, soit le véhicule de la crème deux fois par jour sur les zones affectées des mains et des poignets pendant 16 semaines. Tous les patients ayant terminé les deux études pivots étaient éligibles pour participer à l'étude d'extension à long terme DELTA 3.
Critères d'évaluation
Dans les études DELTA 1 et DELTA 2, le critère d'évaluation principal était la proportion de patients ayant répondu au traitement selon l'évaluation globale de l'investigateur pour l'ECM (IGA-CHE Treatment Success - IGA-CHE TS), définie comme l'obtention d'un score IGA-CHE de 0 (aucune lésion) ou de 1 (presque aucune lésion : seul un érythème à peine perceptible) avec une amélioration d'au moins 2 points entre l'inclusion et la semaine 16. L'instrument IGA-CHE évalue la sévérité de la maladie globale du sujet et se base sur une échelle de 5 points allant de 0 (aucune lésion) à 4 (sévère).
Les autres critères d'efficacité comprenaient l'indice de sévérité de l'eczéma des mains (Hand Eczema Severity Index, HECSI) et le HESD à différents moments. L'HECSI évalue la sévérité de six signes cliniques (érythème, infiltration/formation de papules, vésicules, fissures, desquamation et œdème) et l'étendue des lésions sur chacune des cinq régions des mains (doigts, pulpes, paumes des mains, dos des mains et poignets). L'HESD est un instrument de mesure des résultats rapportés par les patients (Patient-Reported Outcomes, PRO) quotidiennement, comprenant 6 items, conçu pour évaluer la sévérité maximale des signes et symptômes de l'ECM (prurit, douleur, fissures, rougeur, sécheresse et desquamation) à l'aide d'une échelle d'évaluation numérique à 11 points.
Caractéristiques à l'inclusion
Dans tous les groupes de traitement de DELTA 1 et DELTA 2, l'âge moyen était de 44,1 ans, 7,6 % des patients étaient âgés de 65 ans ou plus, 64,4 % étaient des femmes, 90,4 % étaient d'origine caucasienne, 3,5 % étaient d'origine asiatique et 0,7 % étaient d'origine africaine. La fréquence de l'ECM par sous-type principal était de 35,9 % pour l'eczéma atopique des mains, de 21,5 % pour l'eczéma hyperkératosique, de 19,6 % pour la dermatite de contact irritative, de 13,9 % pour la dermatite allergique de contact, de 9,1 % pour l'eczéma vésiculaire des mains (dyshidrose bulleuse) et de 0,1 % pour l'urticaire de contact ou la dermatite de contact aux protéines. Dans les études DELTA 1 et DELTA 2, 71,6 % des patients avaient un score IGA-CHE à l'inclusion de 3 (ECM modéré) et 28,4 % des patients avaient un score IGA-CHE à l'inclusion de 4 (ECM sévère). Le score moyen à l'inclusion au questionnaire sur la qualité de vie en dermatologie (Dermatology Life Quality Index, DLQI) était de 12,5, le score HECSI était de 71,6 et le score HESD de 7,1. Les scores moyens HESD pour le prurit et la douleur étaient respectivement de 7,1 et de 6,7.
Réponse clinique
DELTA 1 et DELTA 2
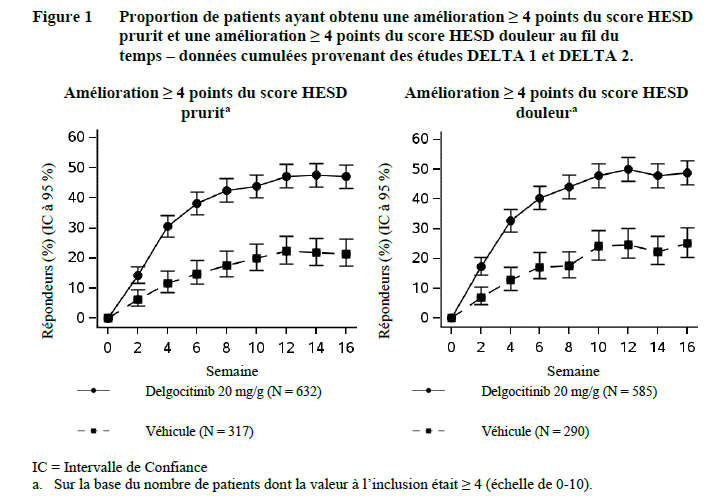
Dans les études DELTA 1 et DELTA 2, une proportion statistiquement plus importante de patients randomisés dans le groupe delgocitinib crème a atteint le critère d'évaluation principal IGA-CHE TS par rapport au véhicule à la semaine 16. Les résultats du critère d'évaluation principal et des critères d'évaluation secondaires les plus pertinents contrôlés pour la multiplicité sont présentés dans le Tableau 3. La Figure 1 illustre la proportion de patients ayant atteint une amélioration ≥ 4 points du score HESD pour le prurit et une amélioration ≥ 4 points du score HESD pour la douleur, au cours du temps, dans les études DELTA 1 et DELTA 2.
Tableau 3 Résultats d'efficacité du delgocitinib à la semaine 16 dans DELTA 1 et DELTA 2
|
|
DELTA 1 |
DELTA 2 |
||
|
|
Delgocitinib (N = 325) |
Véhicule (N = 162) |
Delgocitinib (N = 313) |
Véhicule (N = 159) |
|
IGA-CHE TS, % de répondeursa |
19,7# |
9,9 |
29,1§ |
6,9 |
|
HECSI-90, % de répondeursa, b |
29,5§ |
12,3 |
31,0§ |
8,8 |
|
HECSI-75, % de répondeursa, c |
49,2§ |
23,5 |
49,5§ |
18,2 |
|
HECSI, % de variation moyenne des MC par rapport à l'inclusion (± ET)d |
-56,5§ (± 3,4) |
-21,2 (± 4,8) |
-58,9§ (± 3,2) |
-13,4 (± 4,5) |
|
Amélioration ≥ 4 points du score HESD prurit, % de répondeursa, e |
47,1§ (152/323) |
23,0 (37/161) |
47,2§ (146/309) |
19,9 (31/156) |
|
Amélioration ≥ 4 points du score HESD douleur, % de répondeursa, e |
49,1§ (143/291) |
27,5 (41/149) |
48,6§ (143/294) |
22,7 (32/141) |
|
Amélioration ≥ 4 points du score HESD, % de répondeursa, e |
47,2§ (146/309) |
24,4 (38/156) |
44,5§ (137/308) |
20,9 (32/153) |
#p < 0,01, §p < 0,001
Toutes les valeurs p étaient statistiquement significatives par rapport au véhicule, après ajustement pour la multiplicité.
Abréviations : ET = erreur type ; MC = moindres carrés ; N = nombre de patients dans la population d'analyse totale (tous les patients randomisés et ayant reçu une dose) a. Les données recueillies après l'initiation d'un traitement de secours, l'arrêt définitif du traitement ou les données manquantes ont été considérés comme des non-réponses.
b. Les répondeurs HECSI-90 étaient des patients présentant une amélioration ≥ 90 % du score HECSI par rapport à l'inclusion.
c. Les répondeurs HECSI-75 étaient des patients présentant une amélioration ≥ 75 % du score HECSI par rapport à l'inclusion.
d. Les données recueillies après l'initiation d'un traitement de secours, l'arrêt définitif du traitement ou les données manquantes ont été considérées comme des non-réponses, en appliquant la méthode de la dernière observation rapportée la plus défavorable.
e. Sur la base du nombre de patients dont la valeur à l'inclusion était ≥ 4 (échelle de 0-10).
Dans les études DELTA 1 et DELTA 2, une proportion statistiquement plus élevée de patients traités par le delgocitinib crème a atteint la réussite du traitement IGA-CHE TS et une amélioration ≥ 4 points du score HESD dès la semaine 4, comparativement au véhicule. Une proportion statistiquement plus importante de patients traités par le delgocitinib crème a obtenu un score HECSI-75 à la semaine 8 par rapport au véhicule.
Résultats supplémentaires liés à la qualité de vie/rapportés par les patients
Dans les études DELTA 1 et DELTA 2, les patients traités par le delgocitinib crème ont présenté une amélioration statistiquement significative sur l'échelle de l'impact de l'eczéma des mains (Hand Eczema Impact Scale, HEIS) entre l'inclusion et la semaine 16, par rapport au véhicule (voir Tableau 4). L'HEIS est une échelle utilisée pour évaluer l'impact, tel que perçu par le patient, sur ses activités quotidiennes (utiliser du savon/des produits de nettoyage, effectuer des travaux ménagers nécessitant de se mouiller les mains, se laver, gêne, frustration, sommeil, travailler et tenir ou saisir des objets). 9 items sont évalués sur une échelle à 5 points, où 0 correspond à « pas du tout » et 4 à « extrêmement », et le score HEIS est ensuite calculé en faisant la moyenne de ces 9 items.
Dans les études DELTA 1 et DELTA 2, des améliorations statistiquement significatives de la qualité de vie liée à la santé, mesurée par le DLQI, ont été observées chez les patients traités par delgocitinib par rapport au véhicule à la semaine 16 (voir Tableau 4).
Tableau 4 Résultats liés à la qualité de vie/rapportés par les patients sur le delgocitinib à la semaine 16 dans DELTA 1 et DELTA 2
|
|
DELTA 1 |
DELTA 2 |
|||
|
|
Delgocitinib (N = 325) |
Véhicule (N = 162) |
Delgocitinib (N = 313) |
Véhicule (N = 159) |
|
|
HEIS, variation moyenne des MC par rapport à l'inclusion (± ET)a |
-1,46§ (± 0,05) |
-0,82 (± 0,08) |
-1,45§ (± 0,06) |
-0,64 (± 0,08) |
|
|
HEIS restrictions des activités quotidiennes, variation moyenne des MC par rapport à l'inclusion (± ET)a, b |
-1,46§ (± 0,06) |
-0,86 (± 0,08) |
-1,48§ (± 0,06) |
-0,66 (± 0,08) |
|
|
Amélioration ≥ 4 points du score DLQI, % de répondeursc, d |
74,4§ (227/305) |
50,0 (74/148) |
72,2§ (216/299) |
45,8 (70/153) |
|
|
§p < 0,001 |
|
|
|||
Toutes les valeurs p étaient statistiquement significatives par rapport au véhicule, après ajustement pour la multiplicité.
Abréviations : ET = erreur type ; MC = moindres carrés ; N = nombre de patients dans la population d'analyse totale (tous les patients randomisés et ayant reçu une dose) a. Les données recueillies après l'initiation d'un traitement de secours, l'arrêt définitif du traitement ou les données manquantes ont été considérées comme des non-réponses, en appliquant la méthode de la dernière observation rapportée la plus défavorable.
b. L'HEIS évaluant les restrictions des activités quotidiennes mesure la capacité du patient à utiliser du savon/des produits de nettoyage, à effectuer des travaux ménagers et à se laver. Le score HEIS pour les restrictions des activités quotidiennes est calculé en faisant la moyenne des 3 items.
c. Les données recueillies après l'initiation d'un traitement de secours, l'arrêt définitif du traitement ou les données manquantes ont été considérés comme des non-réponses.
d. Sur la base du nombre de patients dont la valeur à l'inclusion était ≥ 4.
Étude d'extension (DELTA 3)
Les patients ayant terminé les études DELTA 1 ou DELTA 2 pouvaient participer à une étude d'extension en ouvert de 36 semaines (DELTA 3). Dans l'étude DELTA 3, la sécurité et l'efficacité à long terme du traitement par delgocitinib administré selon les besoins ont été évaluées chez 801 patients. Les patients ont commencé à appliquer le delgocitinib crème deux fois par jour sur les zones affectées lorsque le score IGA-CHE était ≥ 2 (d'intensité légère ou plus sévère) et ont arrêté le traitement lorsque le score IGA-CHE de 0 ou 1 était atteint (aucune lésion ou presque aucune lésion). Les patients entrés dans l'étude DELTA 3 avec un score IGA-CHE de 0 ou 1 sont restés sans traitement jusqu'à la perte de réponse (score IGA-CHE ≥ 2).
Les proportions de patients ayant atteint un score IGA-CHE de 0 ou 1, un score HECSI-75, un score HECSI-90, une amélioration ≥ 4 points du score HESD pour le prurit et une amélioration ≥ 4 points du score HESD pour la douleur après la période de traitement initiale de 16 semaines par le delgocitinib crème, ont été maintenues jusqu'à la semaine 52 avec un traitement administré selon les besoins. Parmi les 560 patients randomisés pour recevoir le traitement par le delgocitinib crème dans les études pivots (DELTA 1 et DELTA 2) et inclus dans l'étude DELTA 3, le nombre moyen de périodes de traitement était de 1,5 (avec un intervalle de 0 à 6). La durée moyenne d'une période de traitement était de 123 jours et le nombre cumulé moyen de jours avec une réponse (jours avec un score IGACHE de 0 ou 1 durant la période de traitement de 36 semaines) était de 46. Le nombre cumulé moyen de jours avec une réponse était de 111 parmi les patients ayant atteint la réussite du traitement IGA-CHE TS à la semaine 16 dans les études pivots.
Parmi les patients randomisés pour recevoir le delgocitinib crème dans les études pivots et ayant atteint IGA-CHE TS à la semaine 16, la durée médiane de la réponse durant la période sans traitement était de 4 semaines, et 28,3 % d'entre eux ont maintenu la réponse pendant au moins 8 semaines. Le délai médian pour retrouver un score IGA-CHE de 0 ou 1 après la reprise du traitement était de 8 semaines. Parmi les patients qui n'avaient pas atteint IGA-CHE TS à la semaine 16 du traitement par delgocitinib dans les études pivots, 48,1 % ont obtenu un score IGA-CHE de 0 ou 1 en continuant le traitement par delgocitinib dans l'étude DELTA 3.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec le delgocitinib dans un ou plusieurs sous-groupes de la population pédiatrique pour le traitement de l'eczéma chronique de la main (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
La pharmacocinétique du delgocitinib crème a été évaluée dans une étude menée auprès de 15 patients adultes âgés de 22 à 69 ans présentant un ECM modéré à sévère. Les patients ont appliqué en moyenne 0,87 g de delgocitinib 20 mg/g, crème sur les zones affectées des mains et des poignets deux fois par jour pendant 8 jours.
Les moyennes géométriques de la concentration plasmatique maximale (Cmax) et de l'aire sous la courbe des concentrations en fonction du temps de 0 à 12 heures (ASC0-12) au jour 8 étaient respectivement de 0,46 ng/mL (écart type géométrique [ETG] de 1,74) et de 3,7 ng*h/mL (ETG de 1,74). L'état d'équilibre a été atteint au jour 8. L'exposition systémique (ASC et Cmax) entre le jour 1 et le jour 8 était similaire.
Après une application deux fois par jour de delgocitinib 20 mg/g, crème dans l'étude DELTA 2, la moyenne géométrique de la concentration plasmatique, observée entre 2 et 6 heures après l'application au jour 113, était de 48 % inférieure à celle du jour 8 (0,11 ng/mL et 0,21 ng/mL, respectivement).
La biodisponibilité relative du delgocitinib après application topique est d'environ 0,6 % comparée à celle obtenue avec l'administration de comprimés oraux.
Distribution
D'après une étude in vitro, la liaison du delgocitinib aux protéines plasmatiques est comprise entre 22 et 29 %.
Biotransformation
Le delgocitinib n'étant pas fortement métabolisé, le principal constituant plasmatique est le delgocitinib inchangé. Après administration orale, quatre métabolites (formés par oxydation et conjugaison glucuronide) ont été détectés à < 2 % des concentrations plasmatiques moyennes de delgocitinib inchangé. Le métabolisme limité du delgocitinib est principalement assuré par le CYP3A4/5 et, dans une moindre mesure, par le CYP1A1, le CYP2C19 et le CYP2D6.
Études d'interaction in vitro
D'après les données in vitro, le delgocitinib n'inhibe ni n'induit les enzymes du cytochrome P450 et n'inhibe pas les systèmes de transport tels que les transporteurs d'anions organiques (OAT), les polypeptides de transport d'anions organiques (OATP), les transporteurs de cations organiques (OCT), la P-glycoprotéine (P-gp), la protéine de résistance au cancer du sein (BCRP) ou les protéines d'extrusion multi-médicaments et toxines (MATE) à des concentrations cliniquement pertinentes.
Le delgocitinib est un substrat de la P-glycoprotéine (P-gp) et un substrat faible du transporteur de cations organiques humain 2 (OTC2) et du transporteur d'anions organiques humain 3 (OAT3).
Élimination
Le delgocitinib est principalement éliminé par excrétion rénale, car environ 70 à 80 % de la dose totale administrée par voie orale ont été retrouvés sous forme inchangée dans les urines.
Après une application topique répétée de delgocitinib crème, la demi-vie moyenne du delgocitinib a été estimée à 20,3 heures.
Populations particulières
Insuffisance hépatique
Aucune étude formelle n'a été réalisée sur le delgocitinib crème chez les patients présentant une insuffisance hépatique.
En raison de l'exposition systémique minimale du delgocitinib appliqué par voie topique et de son métabolisme limité, il est peu probable que des modifications de la fonction hépatique aient un effet sur l'élimination du delgocitinib (voir rubrique Posologie et mode d'administration).
Insuffisance rénale
Les paramètres pharmacocinétiques du delgocitinib ont été analysés chez 96 patients présentant une insuffisance rénale légère ou modérée (DFGe de 30 à 89 mL/min/1,73 m2) dans l'étude DELTA 2. Aucune différence cliniquement pertinente n'a été observée dans la pharmacocinétique des patients présentant une insuffisance rénale légère ou modérée par rapport à la population globale de l'étude. Il est peu probable que des altérations de la fonction rénale aient un impact cliniquement significatif sur l'exposition au delgocitinib, en raison de l'exposition systémique minimale après administration topique (voir rubrique Posologie et mode d'administration).
Anzupgo n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, de génotoxicité, de phototoxicité, de tolérance locale, de sensibilisation de la peau et de toxicité juvénile, n'ont pas révélé de risque particulier pour l'homme. Dans les études de toxicologie en administration répétée, des effets ont été observés uniquement à des expositions considérées comme suffisamment supérieures à l'exposition maximale observée chez l'homme après une application topique.
Carcinogénicité
Dans une étude de carcinogénicité cutanée de 2 ans chez la souris, aucune lésion néoplasique locale ou systémique liée au médicament n'a été observée (à des expositions allant jusqu'à environ 600 fois l'exposition humaine sur la base de l'ASC).
Fertilité et développement embryonnaire précoce
Le delgocitinib administré par voie orale n'a pas eu d'effets sur la fertilité, quelle que soit la dose évaluée chez les rats mâles (exposition environ 1 700 fois supérieure à l'exposition humaine). Chez les rats femelles, l'administration orale du delgocitinib a entraîné des effets sur la fertilité (baisse de l'indice de fertilité, réduction du nombre de corps jaunes et d'implantations) à des expositions environ 5 800 fois supérieures à l'exposition humaine. Des pertes post-implantatoires et une diminution du nombre d'embryons vivants ont été observées à des expositions d'environ 432 et 1 000 fois l'exposition humaine, respectivement.
Développement embryofœtal
L'administration orale du delgocitinib n'a pas entraîné d'effets indésirables sur le fœtus chez le rat ou le lapin à des expositions environ 120 et 194 fois supérieures à l'exposition humaine, respectivement. Aucun effet tératogène n'a été observé à aucune des doses étudiées chez le rat ou le lapin (expositions d'environ 1 400 et 992 fois l'exposition humaine, respectivement).
Chez les rats, une diminution du poids des fœtus et des variations squelettiques ont été observées à des expositions 512 fois supérieures à l'exposition humaine, et une tendance à l'augmentation de la perte post-implantatoire a été observée à des expositions environ 1 400 fois supérieures à l'exposition humaine. Chez les lapins, une augmentation de la perte post-implantatoire, une réduction du nombre de fœtus vivants et une tendance à la diminution du poids des fœtus ont été observées à des expositions environ 992 fois supérieures à l'exposition humaine.
Aucun effet pendant la grossesse n'est attendu dans la mesure où l'exposition systémique au delgocitinib est négligeable. Par mesure de précaution, il est préférable d'éviter l'utilisation du delgocitinib pendant la grossesse (voir rubrique Fertilité, grossesse et allaitement).
Développement pré- et post-natal
L'administration orale du delgocitinib à des rats a entraîné une diminution de la viabilité fœtale et une réduction du poids des petits pendant la période postnatale précoce à des expositions plus de 2 000 fois supérieures à l'exposition humaine. Aucun effet sur les évaluations du comportement et de l'apprentissage, la maturation sexuelle ou les performances reproductives de la progéniture, quelle que soit la dose étudiée, n'a été observé.
Après administration orale à des rates allaitantes, le delgocitinib a été sécrété dans le lait à des concentrations environ trois fois supérieures à celles du plasma.
Aucun effet sur le nouveau-né/nourrisson allaité n'est attendu dans la mesure où l'exposition systémique au delgocitinib de la femme qui allaite est négligeable. Le delgocitinib peut donc être utilisé pendant l'allaitement (voir rubrique Fertilité, grossesse et allaitement).
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Médicament nécessitant une surveillance particulière pendant le traitement
Prescription réservée aux spécialistes et services ALLERGOLOGIE
Prescription réservée aux spécialistes et services DERMATOLOGIE
Crème
Crème blanche à légèrement brune
Tube laminé avec une couche barrière en aluminium et une couche intérieure en polyéthylène basse densité, équipé d'un bouchon à charnière en polypropylène.
Présentations : 1 tube de 60 g.
Chaque gramme de crème contient 20 mg de delgocitinib.
Excipients à effet notoire :
Chaque gramme de crème contient 10 mg d'alcool benzylique (E 1519), 0,2 mg de butylhydroxyanisole (E 320) et 72 mg d'alcool cétostéarylique.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Alcool benzylique (E 1519)
Butylhydroxyanisole (E 320)
Alcool cétostéarylique
Acide citrique monohydraté (E 330)
Édétate disodique
Acide chlorhydrique (E 507) (pour l'ajustement du pH)
Paraffine liquide
Éther cétostéarylique de macrogol
Eau purifiée